
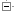
Table of Contents
- Entering a query sequence
- Selecting the species
- Selecting the search tool
- Recent BLAST tickets
- Results
- Configuration options
- References
BLAST [1] and BLAT [2] are sequence similarity search tools that can be used for both DNA and proteins. BLAT is the default tool in Ensembl due to its faster speed. See other differences between BLAT and BLAST on our FAQ page.
How to run BLAST/BLAT
1) Entering a query sequence
Paste in a sequence (the suggested format is FASTA) or upload a sequence as a file. Up to 30 sequences can be added. If inputting multiple species, make sure a header (for example a FASTA header) separates each one.
2) Selecting the species to search against
You can perform multiple similarity searches at once by choosing different genomes and adding them to your list of species.
Click on 'Add/Remove Species' to open the Species Selector box. If you start typing the species name you wish to add, the search box will auto-fill with matches. Selecting these species will add them to your BLAST search. Alternatively, you can click on the species divisions (in green) to browse and select (by checking the boxes) any of the available species in Ensembl. The selected species will appear on the right side of the species selector box, to remove species; click the (-) button on the right of its name. Once you have selected all the species you wish to run through BLAST/BLAT, click apply to return to the query page.
The databases available for similarity searches are DNA and protein target databases.
DNA databases
- Genomic sequence
Repetitve and/or low complexity regions are not masked
- Genomic sequence (hard masked)
Genomic sequences have been run through the RepeatMasker program and repetitive and/or low complexity regions have been masked as Ns
- Genomic sequence (soft masked)
Genomic sequences have been run through the RepeatMasker program and repetitive and/or low complexity regions have been masked as lower case letters
- cDNAs (transcripts/splice variant)
- Ab-initio cDNAs (Genscan/SNAP)
Predictions based on the sequence alone, therefore not supported by experimental evidence
- Ensembl Non-coding RNA genes
Protein databases
- Proteins (GENCODE/Ensembl)
- Ab-initio peptides (Genscan/SNAP)
3) Selecting the search tool
The following options are available:
- BLAT: nucleotide sequences against nucleotide databases
- BLASTN: nucleotide sequences against nucleotide databases
- TBLASTX: translated nucleotide sequences against a translated nucleotide database
- BLASTX: nucleotide sequences against amino acid databases
- BLASTP: amino acid databases against protein databases
- TBLASTN: amino acid databases against nucleotide databases
Pre-Configured Sets
For BLAST searches, you can change the 'Search Sensitivity' from normal to the following:
Near match (to find closer matches- more stringent settings than 'normal')
Short sequences (for short sequences like primers: BLASTN only.)
Distant homologies (to allow lower-scoring pairs to pass through)
Specific parameters for these configurations can be found by expanding the Configuration options. Alternatively, change the configuration options to customise your own BLAST search.
Running the job
You can give a name or description to this BLAST or BLAT search in the Description (optional) field.
Once your parameters are set, click RUN to start the search.
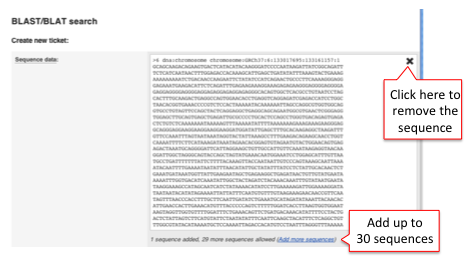
4) Recent BLAST tickets
The table lists jobs that are currently running or recently completed. A ticket ID is assigned to each job and additional information is provided i.e. Analysis, Jobs and Submitted at (date and time). You can customise the table by showing/hiding columns.
The progression of the job gets automatically refreshed every 10 seconds until the job is fully completed.
You can view the results by clicking on the ticket number or on the link View results.
5) Results
The results are displayed in three different sections:
A) Job details
Details of the job include job name, species, search type (e.g. BLAT), sequence, query and database types and configuration settings.
Click on the title or (-) to collapse the Job details section.
B) Results table
It can be viewed on the page or downloaded as a file.
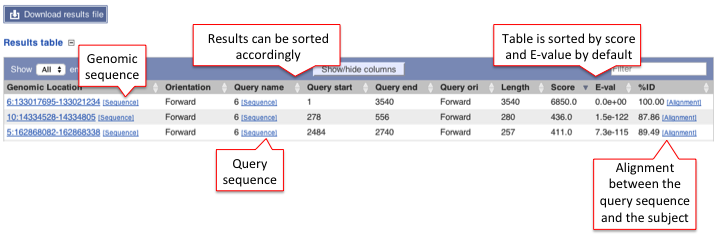
This table lists all hits in order of high to low score (and E-value) but it can be customised to show/hide columns. The results can be sorted by any parameters available in the table.
Hover over the links provided in the results table and click on them to get:
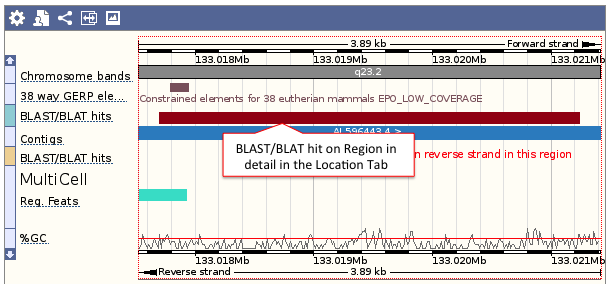
- Genomic location: shows the BLAST/BLAT hit on the Region in detail view in the Location tab of the Ensembl Genome Browser. The BLAST/BLAT hit will appear as a red bar along the genome. You may want to click on the red bar to view a summary of the search, including E-Value, %ID, etc.
- Sequence: shows the genomic sequence or query sequence
- Alignment: shows the BLAST/BLAT alignment
Click on the title or (-) to collaspe the Results table.
C) HSP distribution on genome
High-scoring segment pair (HSP) is a local alignment with no gaps that achieves one of the highest alignment scores in a given search. It corresponds to the matching region between the query and the database hit sequence.
The HSP distribution can be visualised on the karyotype (if the karyotype is available for a given species) and the hits are represented as arrows (the best hit is represented in a box).
Click on the arrows for a pop-up window with a summary of BLAST/BLAT hits such as Genomic location (bp), Score, E-value, etc for all target features available. Links to the Alignment (A), Query sequence (S) and Genomic sequence (G) are also available.

Click on the title or (-) to collaspe the karyotype image.
D) HSP distribution on query sequence
The HSP distribution can be visualised on the query, which is shown as a chain of black and white boxes. Fragments of the query sequence that hit other places in the genome are shown as red boxes (click on those for more information). Usually these fragments are small (they vary between 100-200 nt) and map to various locations. These sequences are of low complexity, such as repetitive sequences.
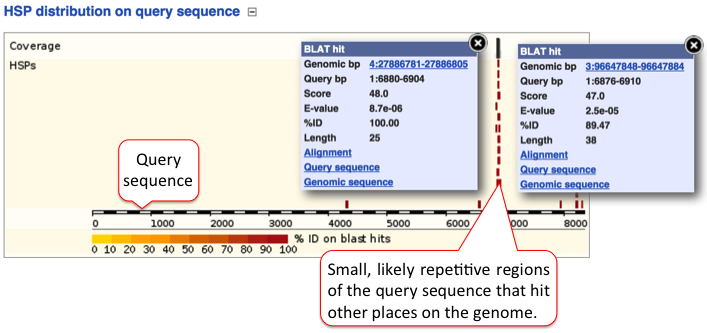
Click on the title or (-) to collaspe the query sequence image.
6) Configuration Options
General options
A) Maximum number of hits to report:
Number of database hits that are displayed. The actual number of alignments may be greater than this. It varies from 10 to 5000 to 100000 and the default is 100.
B) Maximum E-value for reported alignments:
Number of hits reported that contain lover than the E-values selected. It varies from 1e-200 to 1000 and the defatul value is 1e-1.
C) Word size for seeding alignments:
This option is available for BLAST searches only, not BLAT. It is the length of the seed that initiates and alignment between the query and the target sequences. It varies from 2 to 15 and the default is 11 (nucleotides) for DNA and 3 (residues) for protein.
7) References
1) BLAST. Joseph Bedell, Ian Korf and Mark Yandell [OReilly & Associates, 2003]
2) Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002 Apr;12(4):656-64
Sorry, this tutorial has not yet been added to our channel.